Preface
This section studies some first order nonlinear ordinary differential equations describing the time evolution (or “motion”) of spreading some deceases, including HIV, SIR, SEIR, and others.
HIV Models
The interaction of the HIV-1 virus with the body's immune system can be modeled by a system of differential equations similar to a predator-prey system. After an individual is infected with the HIV-1 virus, the amount of the virus in the bloodstream rises dramatically and the person will often suffer from flu-like symptoms. However, these symptoms will disappear after a period of weeks or months as the body begins to manufacture antibodies against the virus. Tests have been developed to determine the presence of HIV-1 antibodies. If a individual has such antibodies, then they are said to be HIV-1 positive. Once infected with the HIV-1 virus, it can be years before an HIV-positive patient exhibits the full symptoms of AIDS. The body's immune system fights the HIV-1 virus with white blood cells. The CD4-positive T-helper cells, a specific type of white blood cell, is especially important since it helps other cells fight the virus. However, the HIV-1 virus can destroy CD4-positive T-helper cells.
We can develop a system of differential equations to better understand the dynamics of the HIV-1 virus. Let V=V(t) be the population of the HIV-1 virus at time t. We will assume that the virus concentration is governed by the following differential equation,
Now let us consider a model for the concentration of T=T(t) of (uninfected) CD4-positive T-helper cells,
The above equation has two equilibrium solutions:
Like the influenza virus, the HIV-1 virus is an RNA virus. An RNA virus cannot reproduce on its own and must use the DNA from a host cell. To do this, the virus attaches itself to a CD4-positive T-helper cell and injects its RNA into the cell. This way the virus can use the T-cell's DNA to replicate itself using a process called reverse transcription, where a DNA copy of the virus's RNA is made. New virus particles are created, and the T cell eventually bursts releasing the virus into the body. If we let I be the concentration of infected T-cells, we can model this process with the following system of equations,
The above system has one nontrivial critical point:
Outer[D, f, x] /; Equal @@ (Dimensions /@ {f, x})
a = {s + p*T*(1 - T/tmax) - dt*T - k*T*V, k*T*V - d*i, n*d*i - c*V}
b = {T, i, V}
JacobianMatrix[a, b] // MatrixForm
One class of drugs that HIV infected patients receive are reverse transcriptase (RT) inhibitors. RT inhibitors block the action of reverse transcription and prevent the virus from duplicating. If one could find the perfect RT inhibitor, then k=0 and our system becomes
Unfortunately, no one has discovered a perfect RT inhibitor, so we will need to modify the system to account for the effectiveness of the RT inhibitor. We can accomplish this by adding an effectiveness factor 1-η to the k VT term. Our system now becomes
If η=1, then the RT inhibitor is completely effective. On the other hand, if η=0, then the RT inhibitor is completely ineffective. We now have a model for how the HIV-1 virus interacts with the immune system. Researchers can use data to estimate the parameters and see exactly what types of solutions are possible.
| Symbol | Meaning | Value |
|---|---|---|
| a | average per capita birth rate of foxes | 1 yr-1 |
| b | average per capita intrinsic death rate | 0.5 yr-1 |
| β | rabies transmission coefficient | 79.67 km² yr-1 |
| σ | 1/σ is the average latent period (∼ 28 to 30 days) | 13 yr-1 |
| α | death rate of rabid foxes (average life expectancy ∼ 5 days) | 73 yr-1 |
| γ | &gammalN represents increased death rate when N is large enough to deplete the food supply | 0.1--5 km²yr-1 |
SIR and SIRS Models
This topic describes the differential equations that govern the classic deterministic SIR and SIRS compartmental models and describes how to configure EMOD, an agent-based stochastic model, to simulate an SIR/SIRS epidemic. In SIR models, individuals in the recovered state gain total immunity to the pathogen; in SIRS models, that immunity wanes over time and individuals can become reinfected. The EMOD generic simulation uses an SEIR-like disease model by default. You can modify the default SEIR model to an SIR model by turning off the incubation period.
As the first step in the modeling process, we identify the independent and dependent variables. The independent variable is time t, measured in days. We consider two related sets of dependent variables. The first set of dependent variables counts people in each of the groups, each as a function of time:
- S = S(t) is the number of susceptible individuals, s the number of susceptible individuals,
- I = I(t) is the number of infected individuals, and
- R = R(t) is the number of recovered individuals.
- s = s(t) = S(t)/N is the susceptible fraction of the population,
- i = i(t) = I(t)/N is the infected fraction of the population, and
- r(t) = R(t)/N is the recovered fraction of the population.
The resulting differential equations for the model are
Tumor-immune Models
A tumor, an abnormal growth of body tissue, may be malignant or benign. A malignant tumor can turn into cancer, which is difficult to cure once metastasized. With the immune system, the host usually controls the growth and division of tumor cells to prevent from becoming cancerous. In this process, the interaction between tumors and immune system is a complex phenomenon difficult to be understood completely. There are known many mathematical models to predict this interaction.
It is known that tumor cells can biochemically stimulate the production of immune cells (commonly called effector cells) such as cytotoxic T-cells, macrophages, and natural killer cells. Effector cells are cytotoxic to tumor cells and will retard the growth of tumor cells. Therefore, the interaction between tumor cells and effector cells can be regarded as an analog of the "predator-prey" interaction, where the predator is effector cells and the prey is tumor cells. On the other hand, effector cells can be neutralized by tumor cells, which means that the two types of cells are "competitive". The coexistence of these two relationships leads to the complexity of the dynamic behavior of resulting models.
The interaction between cells and effector cells are determined by three major factors: the malignant potential of a tumor, the tumor's antigenicity, and the immune response of the host. The malignant potential of a tumor represents its ability to metastasize, escape, and destroy the immune system. The degree of antigenicity of a tumor is defined as the initial size of the effector cell population that can be stimulated upon introduction of the antigen, which is associated with the tumor. It varies greatly from cancer to cancer and from patient to patient. Larger values represent tumor cells that present a well recognized antigen while small values represent tumor cells that present a weak antigen. The tumor response reflects the suppression of tumor growth by the host's immune system, where effector cells are recruited due to the presence of tumor cells. The recruitment of effector cells corresponding to the presence of tumor cells depends on tumor's antigenicity of tumor cells. The classical tumor-immune interaction models were first proposed by Kuznetsov et al. (1994) and Kirschner and Panetta (1998).
Following Li at al. (2021), we consider the following (normalized) model
Disregarding the last term in second equation of \eqref{EqTumor.1}, we get
Obviously, system \eqref{EqTumor.1} always has equilibrium at P₀(s, 0), which is referred to as the tumor-free equilibrium, which belongs to the boundary of Ω. Other equilibrium points must be found from the system of equations
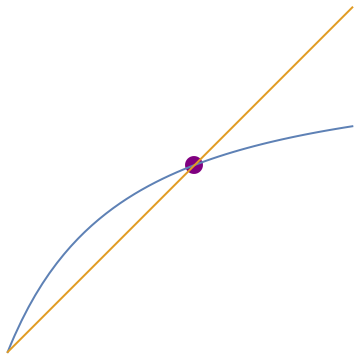
|
f[t_] = 1 - (1 + 0.5*t)/(1 + t)/(1 + 2*t);
plot=Plot[{f[t], t}, {t, 0, 1.2}, PlotStyle -> Thick]; point = Graphics[{PointSize[0.05], Purple, Point[{0.651, 0.651}]}]; Show[point, plot]
NSolve[f[t] == t, t]
{{t -> -1.15139}, {t -> 0.651388}, {t -> 0.}}
|
- Kirschner, D., Panetta, J.C., Modeling immunotherapy of the tumor-immune interaction, Journal of Mathematical biology, 1998, 37, pp. 235--252.
- Kuznetsov, V.A., Makalkin, I.A., Taylor, M.A., Perelson, A.S., Nonlinear dynamics of immunogenic tumors: parameter estimation and global bifurcation analysis, Journal of Mathematical biology, 1994, 56, pp. 295--321.
- Li, J., Xie, X., Chen, Y., Zhang, D., Complex dynamics of a tumor-immune system with antigenicity, Applied Mathematics and Computation, 2021, 400, 126052.
- Bjørnstad, O.N., Shea, K., Krzywinski, M., The SEIRS model for infectious disease dynamics, Nature Methods, 2020, volume 17, pages 557–558. https://doi.org/10.1038/s41592-020-0856-2
- SIR and SIRS Models, IDM
- Smith, D. and Moore, L., The SIR Model for Spread of Disease - The Differential Equation Model, The American Mathematical Monthly,
- Yano, T.K., Makinde, O.D., Malonza, D.M., Modelling Childhood Disease Outbreak in a Community with Inflow of Susceptible and Vaccinated New-born, Global Journal of Pure and Applied Mathematics, 2016, Volume 12, Number 5, pp. 3895-3916.
Return to Mathematica page
Return to the main page (APMA0340)
Return to the Part 1 Matrix Algebra
Return to the Part 2 Linear Systems of Ordinary Differential Equations
Return to the Part 3 Non-linear Systems of Ordinary Differential Equations
Return to the Part 4 Numerical Methods
Return to the Part 5 Fourier Series
Return to the Part 6 Partial Differential Equations
Return to the Part 7 Special Functions